A New Type of Na(+)-Driven ATP Synthase Membrane Rotor with a Two-Carboxylate Ion-Coupling Motif.
Schulz, S., Iglesias-Cans, M., Krah, A., Yildiz, O., Leone, V., Matthies, D., Cook, G.M., Faraldo-Gomez, J.D., Meier, T.(2013) PLoS Biol 11: 01596
- PubMed: 23824040
- DOI: https://doi.org/10.1371/journal.pbio.1001596
- Primary Citation of Related Structures:
3ZK1, 3ZK2 - PubMed Abstract:
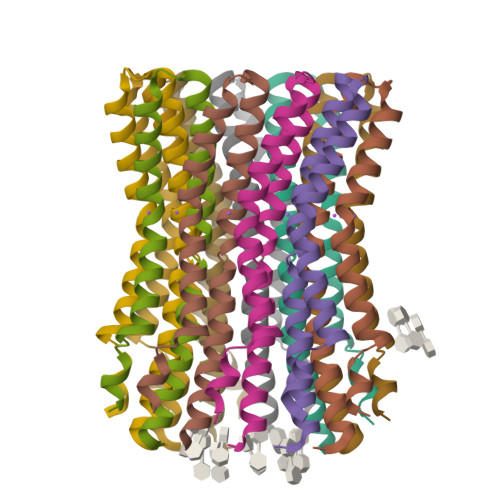
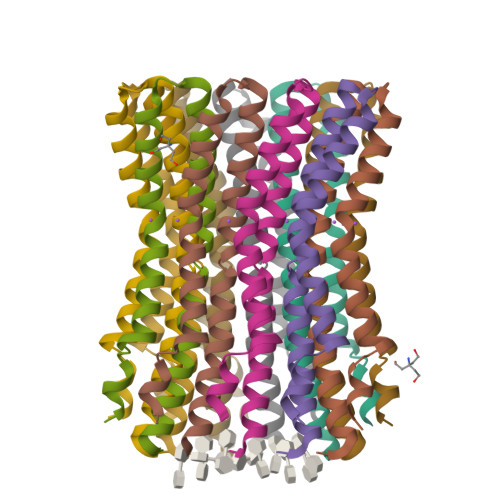
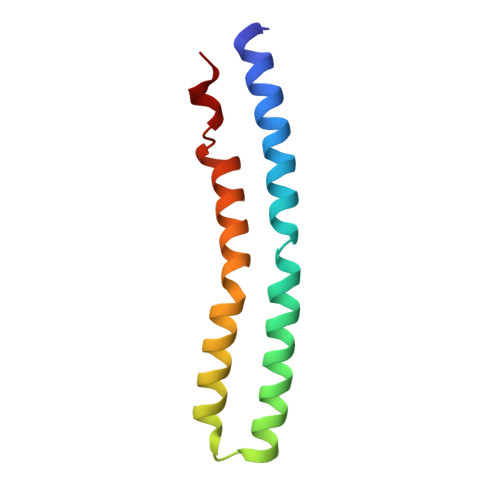
The anaerobic bacterium Fusobacterium nucleatum uses glutamate decarboxylation to generate a transmembrane gradient of Na⁺. Here, we demonstrate that this ion-motive force is directly coupled to ATP synthesis, via an F₁F₀-ATP synthase with a novel Na⁺ recognition motif, shared by other human pathogens. Molecular modeling and free-energy simulations of the rotary element of the enzyme, the c-ring, indicate Na⁺ specificity in physiological settings. Consistently, activity measurements showed Na⁺ stimulation of the enzyme, either membrane-embedded or isolated, and ATP synthesis was sensitive to the Na⁺ ionophore monensin. Furthermore, Na⁺ has a protective effect against inhibitors targeting the ion-binding sites, both in the complete ATP synthase and the isolated c-ring. Definitive evidence of Na⁺ coupling is provided by two identical crystal structures of the c₁₁ ring, solved by X-ray crystallography at 2.2 and 2.6 Å resolution, at pH 5.3 and 8.7, respectively. Na⁺ ions occupy all binding sites, each coordinated by four amino acids and a water molecule. Intriguingly, two carboxylates instead of one mediate ion binding. Simulations and experiments demonstrate that this motif implies that a proton is concurrently bound to all sites, although Na⁺ alone drives the rotary mechanism. The structure thus reveals a new mode of ion coupling in ATP synthases and provides a basis for drug-design efforts against this opportunistic pathogen.
Organizational Affiliation:
Department of Structural Biology, Max Planck Institute of Biophysics, Frankfurt am Main, Germany.