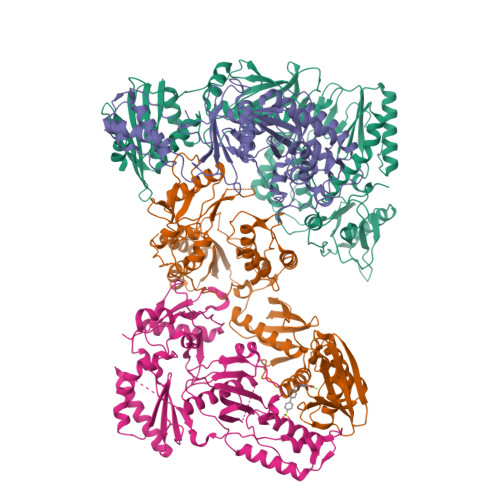
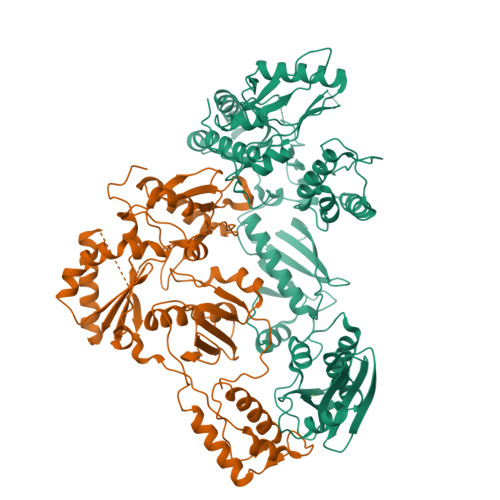
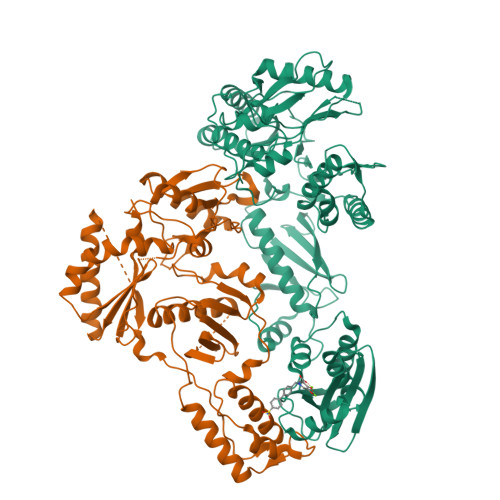
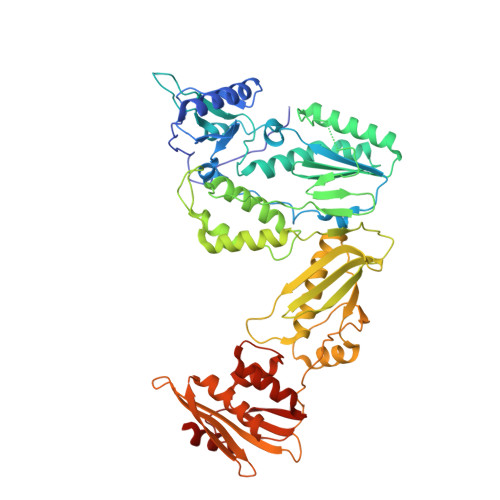
Design, Synthesis, and Biological Evaluations of Hydroxypyridonecarboxylic Acids as Inhibitors of HIV Reverse Transcriptase Associated RNase H.
Kankanala, J., Kirby, K.A., Liu, F., Miller, L., Nagy, E., Wilson, D.J., Parniak, M.A., Sarafianos, S.G., Wang, Z.(2016) J Med Chem 59: 5051-5062
- PubMed: 27094954
- DOI: https://doi.org/10.1021/acs.jmedchem.6b00465
- Primary Citation of Related Structures:
5J1E - PubMed Abstract:
Targeting the clinically unvalidated reverse transcriptase (RT) associated ribonuclease H (RNase H) for human immunodeficiency virus (HIV) drug discovery generally entails chemotypes capable of chelating two divalent metal ions in the RNase H active site. The hydroxypyridonecarboxylic acid scaffold has been implicated in inhibiting homologous HIV integrase (IN) and influenza endonuclease via metal chelation. We report herein the design, synthesis, and biological evaluations of a novel variant of the hydroxypyridonecarboxylic acid scaffold featuring a crucial N-1 benzyl or biarylmethyl moiety. Biochemical studies show that most analogues consistently inhibited HIV RT-associated RNase H in the low micromolar range in the absence of significant inhibition of RT polymerase or IN. One compound showed reasonable cell-based antiviral activity (EC50 = 10 μM). Docking and crystallographic studies corroborate favorable binding to the active site of HIV RNase H, providing a basis for the design of more potent analogues.
Organizational Affiliation:
Center for Drug Design, Academic Health Center, University of Minnesota , Minneapolis, Minnesota 55455, United States.