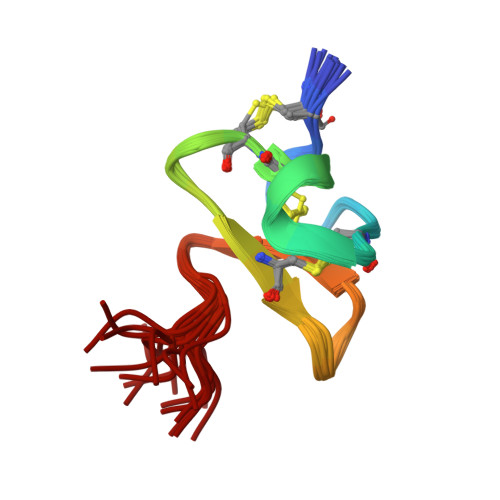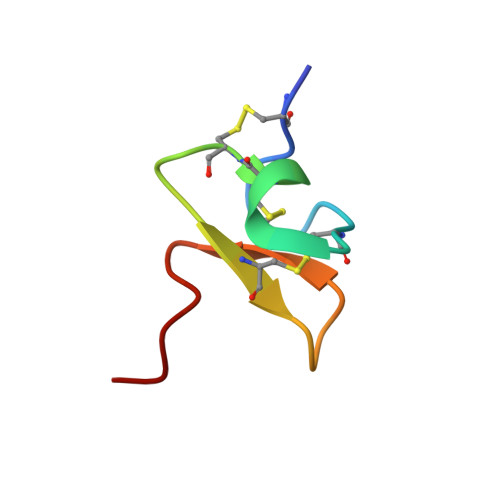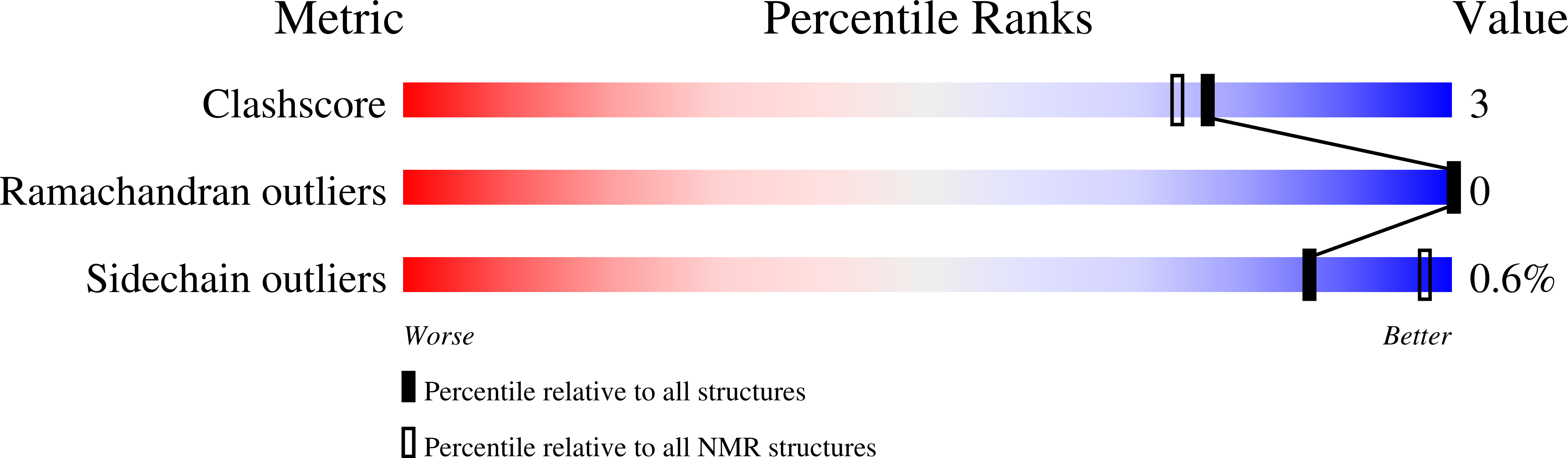
Pharmacological characterisation of the highly Na V 1.7 selective spider venom peptide Pn3a.
Deuis, J.R., Dekan, Z., Wingerd, J.S., Smith, J.J., Munasinghe, N.R., Bhola, R.F., Imlach, W.L., Herzig, V., Armstrong, D.A., Rosengren, K.J., Bosmans, F., Waxman, S.G., Dib-Hajj, S.D., Escoubas, P., Minett, M.S., Christie, M.J., King, G.F., Alewood, P.F., Lewis, R.J., Wood, J.N., Vetter, I.(2017) Sci Rep 7: 40883-40883
- PubMed: 28106092
- DOI: https://doi.org/10.1038/srep40883
- Primary Citation of Related Structures:
5T4R - PubMed Abstract:
Human genetic studies have implicated the voltage-gated sodium channel Na V 1.7 as a therapeutic target for the treatment of pain. A novel peptide, μ-theraphotoxin-Pn3a, isolated from venom of the tarantula Pamphobeteus nigricolor, potently inhibits Na V 1.7 (IC 50 0.9 nM) with at least 40-1000-fold selectivity over all other Na V subtypes. Despite on-target activity in small-diameter dorsal root ganglia, spinal slices, and in a mouse model of pain induced by Na V 1.7 activation, Pn3a alone displayed no analgesic activity in formalin-, carrageenan- or FCA-induced pain in rodents when administered systemically. A broad lack of analgesic activity was also found for the selective Na V 1.7 inhibitors PF-04856264 and phlotoxin 1. However, when administered with subtherapeutic doses of opioids or the enkephalinase inhibitor thiorphan, these subtype-selective Na V 1.7 inhibitors produced profound analgesia. Our results suggest that in these inflammatory models, acute administration of peripherally restricted Na V 1.7 inhibitors can only produce analgesia when administered in combination with an opioid.
Organizational Affiliation:
IMB Centre for Pain Research, Institute for Molecular Bioscience, 306 Carmody Rd (Building 80), The University of Queensland, St Lucia, Queensland, 4072, Australia.