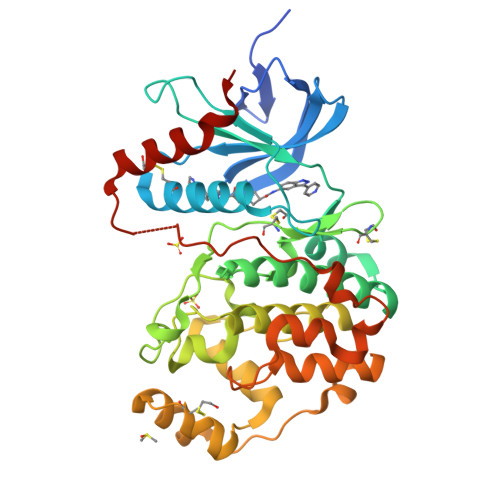
Fragment-Based Discovery of a Potent, Orally Bioavailable Inhibitor That Modulates the Phosphorylation and Catalytic Activity of ERK1/2.
Heightman, T.D., Berdini, V., Braithwaite, H., Buck, I.M., Cassidy, M., Castro, J., Courtin, A., Day, J.E.H., East, C., Fazal, L., Graham, B., Griffiths-Jones, C.M., Lyons, J.F., Martins, V., Muench, S., Munck, J.M., Norton, D., O'Reilly, M., Palmer, N., Pathuri, P., Reader, M., Rees, D.C., Rich, S.J., Richardson, C., Saini, H., Thompson, N.T., Wallis, N.G., Walton, H., Wilsher, N.E., Woolford, A.J., Cooke, M., Cousin, D., Onions, S., Shannon, J., Watts, J., Murray, C.W.(2018) J Med Chem 61: 4978-4992
- PubMed: 29775310
- DOI: https://doi.org/10.1021/acs.jmedchem.8b00421
- Primary Citation of Related Structures:
6G8X, 6G91, 6G92, 6G93, 6G97, 6G9A, 6G9D, 6G9H, 6G9J, 6G9K, 6G9M, 6G9N, 6GDM, 6GDQ, 6GE0 - PubMed Abstract:
Aberrant activation of the MAPK pathway drives cell proliferation in multiple cancers. Inhibitors of BRAF and MEK kinases are approved for the treatment of BRAF mutant melanoma, but resistance frequently emerges, often mediated by increased signaling through ERK1/2. Here, we describe the fragment-based generation of ERK1/2 inhibitors that block catalytic phosphorylation of downstream substrates such as RSK but also modulate phosphorylation of ERK1/2 by MEK without directly inhibiting MEK. X-ray crystallographic and biophysical fragment screening followed by structure-guided optimization and growth from the hinge into a pocket proximal to the C-α helix afforded highly potent ERK1/2 inhibitors with excellent kinome selectivity. In BRAF mutant cells, the lead compound suppresses pRSK and pERK levels and inhibits proliferation at low nanomolar concentrations. The lead exhibits tumor regression upon oral dosing in BRAF mutant xenograft models, providing a promising basis for further optimization toward clinical pERK1/2 modulating ERK1/2 inhibitors.
Organizational Affiliation:
Astex Pharmaceuticals , 436 Cambridge Science Park , Cambridge , CB4 0QA , U.K.